Publications
Preprints
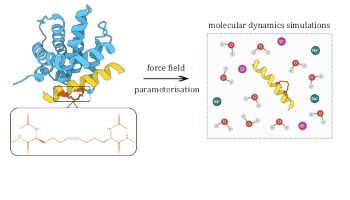
Atomic Environment Aware Lennard-Jones Parameterization for Electrostatic Embedding ML/MM Simulations
João Morado, Kirill Zinovjev, Lester O. Hedges, Julien Michel, Daniel J. Cole,
[ chemRxiv
 ]
]
Quantum Elastic Network Models and their Application to Graphene
Ioannis Kolotouros, Adithya Sireesh, Stuart Ferguson, Sean Thrasher, Petros Wallden, Julien Michel
[ arXiv ]

Adiabatic-Inspired Hybrid Quantum-Classical Methods for Molecular Ground State Preparation
Sean Thrasher, Ioannis Kolotouros, Julien Michel, Petros Wallden
[ arXiv ]
Stapline: Development of a Force Field Library for Stapled Peptide Residues
Evangelia Notari, Marie T. J. Bluntzer, Julien Michel, Alison N. Hulme
[ chemRxiv ]
Journal Publications
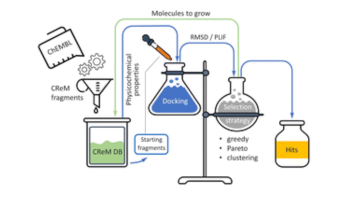
CReM-dock: de novo design of synthetically feasible structures guided by molecular docking
Guzel Minibaeva, Haolin Du, Finlay Clark, Julien Michel, Pavel Polishchuk
Digital Discovery, 5, 2271-2291, 2026 doi:10.1039/D6DD00131A
Computational Design, Synthesis, and Evaluation of Stapled Peptide-Based Antagonists of the CGRP Receptor
Adam L. Schofield, Evangelia Notari, Mária Rožňovcová, Kathryn W. Cox, Vera D’Aloisio, Christian Steuer, Julien Michel, Graeme S. Cottrell, Christopher R. Coxon
J. Med. Chem., 69, 6, 7142–7159, 2026 doi:10.1021/acs.jmedchem.5c03445

A Computational Community Blind Challenge on Pan-Coronavirus Drug Discovery Data
Hugo MacDermott-Opeskin, Jenke Scheen, Cas Wognum, Joshua T. Horton, Devany West, Alexander Matthew Payne, Maria A. Castellanos, Sean Colby, Edward Griffen, David Cousins, Jessica Stacey, Lauren Reid, Jasmin Cara Aschenbrenner, Daren Fearon, Blake Balcomb, Peter Marples, Charles W.E. Tomlinson, Ryan Lithgo, Max Winokan, Haim Barr, Noa Lahav, Michael Lavi, Shirley Duberstein, Galit Cohen, Gwendolyn Fate , Bruce Lefker, Ralph Robinson, Tamas Szommer, Nick Lynch, Kate Huddleston, Mallory Tollefson, Cynthia Xu, Jonny Hsu, Julien St-Laurent, Honore Etsmoberg, Lu Zhu, Andrew Quirke, Mohamed Iliyas Abdul Haleem, Irfan Alibay, Gunjan Baid, Benjamin Birnbaum, Kevin P. Bishop, Hugo Bohorquez, Ashmita Bose, C. J. Brown, Jackson Burns, Lianjin Cai, Ruel Cedeno, Stephane de Cesco, Vladimir Chupakhin, Finlay Clark, Daniel J. Cole, Carles Corbi-Verge, Muhammad Danial, Alec Davi, Wim Dehaen, Niklas Piet Doering , Alexis Dougha, Marie-Pierre Dréanic, Bryce Eakin, Anatol Ehrlich, Rokas Elijosius, Jozef Fülöp, Anthony Gitter, Kenneth Goossens, Yaowen Gu, Teresa Head-Gordon, Laurent Hoffer, Johan Hofmans, Ellena Jiang, Benjamin Kaminow, Sina Khosravi, Asma Feriel Khoualdi, Eelke Bart Lenselink, Zhirong Liu, Yue Liu, Sijie Liu, Yizhou Ma, Patrick Maher, Imke Mayer, Oscar Mendez-Lucio, Antonia Mey, Julien Michel, Floriane Montanari, Taoyu Niu, Ryusei Ogino, Ashok Palaniappan, Xiaolin Pan, Auro Patnaik, Long-Hung Pham, Luis Pinto, Justin Purnomo, Alexander Rich, Lars Schaaf, Christoph Schran, Rajeev Kumar Singh, Mounika Srilakshmi, Satya Pratik Srivastava, Kunyang Sun, Zhaoxi Sun, Valerij Talagayev, Balamurugan Thirukonda Subramanian Balakrishnan, Ida Titus, Alexandre Tkatchenko, Wojtek Treyde, Giovanni Tricarico, Austin Tripp, Nopsinth Vithayapalert, Yingze Wang, Azmine Toushik Wasi, Steffen Wedig, Gerhard Wolber, Bofei Xu, Weijun Zhou, Frank von Delft, Alpha Lee, Karla Kirkegaard, Peter Sjö, James Fraser, John D. Chodera
J. Chem. Inf. Model., 66, 6, 3129–3149, 2026 doi:10.1021/acs.jcim.5c02106 [ chemRxiv ]
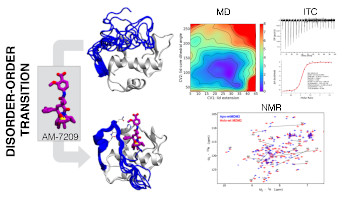
Molecular Driving Force of a Small Molecule- Induced Protein Disorder-Order Transition
Cesar Mendoza-Martinez, Arun A. Gupta, Salomé Llabrés, Paul N. Barlow, Julien Michel
Commun. Chem. 9, 65, 2026 doi:10.1038/s42004-025-01869-5 [ chemRxiv]
Accurate Prediction of Drug Resistance for Intrinsically Disordered Protein Regions
Audrius Kalpokas, Marck Mackey, Julien Michel
J. Chem. Theory Comput., 21, 24, 12497-12507, 2025 doi:10.1021/acs.jctc.5c01648 [ chemRxiv ]
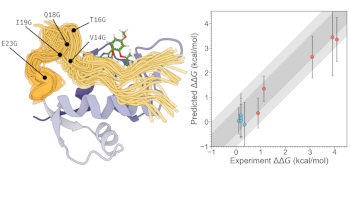
Enhancing Electrostatic Embedding for ML/MM Free Energy Calculations
João Morado, Kirill Zinovjev, Lester O. Hedges, Daniel J. Cole, Julien Michel
J. Chem. Theory Comput., 21, 22, 11805-11819 2025 doi:10.1021/acs.jctc.5c01464 [ chemRxiv ]
Modular and Interoperable Workflows for Benchmarking Alchemical Binding Free Energy Calculation Methodologies
Anna Herz, Maicol Bissaro, Carmen Esposito, Julien Michel
J. Chem. Inf. Model., 65, 19, 10658–10672, 2025 doi:10.1021/acs.jcim.5c01493 [ chemRxiv ]

Elucidation of the Mechanism of Partial Activation of EPAC1 Allosteric Modulators by Markov State Modelling
Adele Hardie, Frederick G Powell, Silvia Lovera, Stephen J. Yarwood, Graeme Barker, Julien Michel
Chem. Sci., 16, 14771-14781, 2025 [ doi:10.1039/D5SC02112J ] [ chemRxiv ]

Optimization of Cyclophilin B-Targeted Tri-vector Inhibitors for Novel MASH Treatments
Maria-Eleni Kouridaki, Jonathan Gillespie, John Robinson, Tanya Mathie, Laura Bain, Duncan McArthur, Angus Morrison, Daniel B. Greenslade, Michail Papadourakis, Kasia Maj, Kate Cameron, Darryl Turner, Scott P. Webster, Martin A. Wear, Dahlia Doughty-Shenton. Alison N. Hulme, Julien Michel
J. Med. Chem., 68,6, 6815-6831, 2025 doi:10.1021/acs.jmedchem.5c00301

Assessment of the Topology and Oligomerisation States of Coiled Coils Using Metadynamics with Conformational Restraints
Evangelia Notari, Christopher Wood, Julien Michel
J. Chem. Theory Comput., 21, 6, 3260-3276, 2025 doi:10.1021/acs.jctc.4c01695 [ chemRxiv ]

Robust Automated Truncation Point Selection for Molecular Simulations
Finlay Clark, Daniel Cole, Julien Michel
J. Chem. Theory Comput., 21, 1, 88-101, 2025 doi:10.1021/acs.jctc.4c01359 [chemRxiv]

Comparison of Methodologies for Absolute Binding Free Energy Calculations of Ligands to Intrinsically Disordered Proteins
Michail Papadourakis, Zoe Cournia, Antonia S. J. S. Mey, Julien Michel
J. Chem. Theory Comput., 20, 21, 9699-9707, 2024 doi:10.1021/acs.jctc.4c00942 [ bioRxiv]

Automated Adaptive Absolute Binding Free Energy Calculations
Finlay Clark, Graeme Robb, Daniel J. Cole, Julien Michel
J. Chem. Theory Comput., 20, 18, 7806-7828, 2024 doi:10.1021/acs.jctc.4c00806 [ chemRxiv]

Sire: An Interoperability Engine for Prototyping Algorithms and Exchanging Information Between Molecular Simulation Programs
Christopher J. Woods, Lester Hedges, Adrian Mulholland, Maturos Malaisree, Paolo Tosco, Hannes H. Loeffler, Miroslav Suruzhon, Matthew Burman, Sofia Bariami, Stefano Bosisio, Gaetano Calabro, Finlay Clark, Antonia S. J. S. Mey, Julien Michel
J. Chem. Phys. , 160, 202503, 2024 doi:10.1063/5.0200458 [ chemRxiv ]

OpenMM 8: Molecular Dynamics Simulation with Machine Learning Potentials
Peter Eastman, Raimondas Galvelis, Raul P. Pelaez, Charlles R. A. Abreu, Stephen E. Farr, Emilio Gallicchio, Anton Gorenko, Michael M. Henry, Frank Hu, Jing Huang, Andres Kramer, Julien Michel, Joshua A. Mitchell, Vijay S. Pande, Joao Rodrigues, Jaime Rodriguez-Guerra, Andrew C. Simmonett, Jason Swails, Ivy Zhang, John. D. Chodera, Gianni De Fabritiis, Thomas E. Markland
J. Phys. Chem. B 128(1), 109-116, 2024 doi:10.1021/acs.jpcb.3c06662 [ arXiv ]

A Suite of Tutorials for the BioSimSpace Framework for Interoperable Biomolecular Simulation
Hedges, L. O. ; Bariami, S. ; Burman, M. ; Clark, F. ; Cossins, B. P. ; Hardie, A. ; Herz, A. M. ; Lukauskis, D. ; Mey, A. S. J. S. ; Michel, J. ; Scheen, J. ; Suruzhon, M. ; Woods, C. J. ; Wu, Z.
Living J. Comp. Mol. Sci. 5 (1), 2023 doi:10.33011/livecoms.5.1.2375 [ github ]
Alchemical Free Energy Workflows for the Computation of Protein-Ligand Binding Affinities
Herz, A. M. ; Kellici, T. ; Morao, I. ; Michel, J.
In: Heifetz, A. (eds) High Performance Computing for Drug Discovery and Biomedicine. Methods mol. biol., vol 2716, 2024
Deconstructing Allostery: Computational Assessment of the Binding Determinants of Allosteric PTP1B Modulators
Hardie, A. ; Cossins, B. P. ; Lovera, S. ; Michel, J.
Commun. Chem. 6, 125, 2023 doi:10.1038/s42004-023-00926-1 [ chemRxiv ]
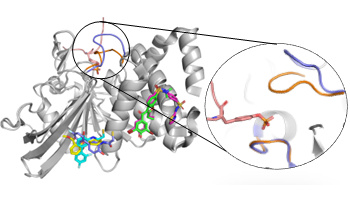
Comparison of Receptor-Ligand Restraint Schemes for Alchemical Absolute Binding Free Energy Calculations
Clark, F. ; Robb, G. ; Cole, D. J. ; Michel, J.
J. Chem. Theory Comput. 19, 12, 3686-3704, 2023 doi:10.1021/acs.jctc.3c00139 [ chemRxiv ]

Data-driven Generation of Perturbation Networks for Relative Binding Free Energy Calculations
Scheen, J. ; Mackey, M. ; Michel, J.
Digital Discovery, 1, 870-885, 2022 doi:10.1039/D2DD00083K [ chemrxiv ]

A fluorogenic probe for granzyme B enables in- biopsy evaluation and screening of response to anticancer immunotherapies
Scott, J. I. ; Mendiva-Tapia, L. ; Gordon, D. ; Barth, N. D. ; Thompson, E. J. ; Cheng, Z. ; Taggart, D. ; Kitamuri, T. ; Blas, A. B. ; Roberts, E. W. ; Juarez-Jimenez, J. ; Michel, J. ; Piet, B. ; de Vries, I. J. ; Verdoes, M. ; Dawson, J. ; Carragher, N. O. ; O'Connor, R. A. ; Akram, A. R. ; Serrels, A. ; Frame, M. ; Vendrell, M.
Nat. Commun. , 13, 2366. 2022, doi:10.1038/s41467-022-29691-w

Energetics of a protein disorder-order transition in small molecule recognition
Mendoza-Martinez, C. ; Papadourakis, M. ; Llabrés, S. ; Gupta, A. A. ; Barlow, P. N. ; Michel, J.
Chem. Sci. , 13, 5220-5229, 2022, doi:10.1039/D2SC00028H [ biorxiv ]
Implementation of the QUBE Force Field for High-Throughput Alchemical Free Energy Calculations
Nelson, L. ; Bariami, S. ; Ringrose, C. ; Horton, J. T. ; Kurdekar, V. ; Mey, A. S. J. ; Michel, J. ; Cole, D. J.
J. Chem. Inf. Model. ,61, 5, 2124-2134, 2021 doi:10.1021/acs.jcim.1c00328 [ chemrxiv ]
Designing Stapled Peptides to Inhibit Protein-Protein Interactions: An Analysis of Successes in a Rapidly Changing Field
Bluntzer, M. T. J. ; O'Connell J. ; Baker, T. S. ; Michel, J. ; Hulme, A. N.
J. Pept. Sci. 113:e24191, 2021 doi:10.1002/pep2.24191

Best Practices for Alchemical Free Energy Calculations
Mey, A. S. J. S ; Allen, N. ; Bruce Macdonald, H. E. B. ; Chodera, J. D. ; Kuhn, M. ; Michel, J. ; Mobley, D. L. ; Naden, L. N. ; Prasad, S. ; Rizzi, A. ; Scheen, J. ; Shirts, M. R. ; Tresadern, G. ; Xu, H.
Living J. Comp. Mol. Sci. 2 (1), 2020 doi:10.33011/livecoms.2.1.18378 [ arxiv ]

Combining Virtual Reality Visualization with Ensemble Molecular Dynamics to Study Complex Protein Conformational Changes
Juárez-Jiménez, J ; Tew, P ; O'Connor M ; Llabres, S ; Sage, R ; Glowacki D ; Michel, J.
J. Chem. Inf Model. 60, 12, 6344-6354, 2020 doi:10.1021/acs.jcim.0c00221 [ chemrxiv ]
A hybrid Alchemical Free Energy/Machine Learning Methodology for the Computation of Hydration Free Energies
Scheen, J ; Wu, W ; Mey, A. S. J. S. ; Tosco, P. ; Mackey, M. ; Michel, J.
J. Chem. Inf Model. 60, 11, 5331-5339, 2020 doi:10.1021/acs.jcim.0c00600 [chemrxiv]
Assessment of Binding Affinity via Alchemical Free Energy Calculations
Kuhn, M ; Firth-Clark, S. ; Tosco, P ; Mey, A. S. J. S. ; Mackey, M. ; Michel, J.
J. Chem. Inf Model. ,60 , 6, 3120–3130, 2020 doi:10.1021/acs.jcim.0c00165 [chemrxiv]
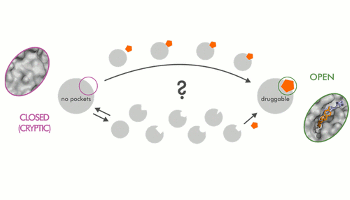
Investigating Cryptic Binding Sites by Molecular Dynamics Simulations
Kuzmanic, A. ; Bowman, G. A. ; Juárez-Jiménez, J. ; Michel, J. ; Gervasio, F. L.
Acc. Chem. Res. , 53, 3, 654-661, 2020 doi:10.1021/acs.accounts.9b00613
The SAMPL6 SAMPLing challenge: Assessing the reliability and efficiency of binding free energy calculations
Rizzi, A. ; Jensen, T. ; Slochower, D. R. ; Aldeghi, M. ; Vytautas, G. ; , Ntekoumes, D. ; Bosisio, S. ; Papadourakis, M. ; Henriksen, N. M. ; de Groot, B. L. ; Cournia, Z. ; Dickson, A. ; Michel, J. ; Gilson, M. K. ; Shirts, M. R. ; Mobley, D. L. ; Chodera, J. D.
J Comput. Aided Mol. Design, 34, 601-633, 2020 doi:10.1007/s10822-020-00290-5 [bioRxiv]

Dynamic design: manipulation of millisecond timescale motions on the energy landscape of Cyclophilin A
Juárez-Jiménez, J.; Gupta, A. A. ; Karunanithy, G.; Mey, A. S. J. S.; Georgiou, C.; Ioannidis, H.; De Simone, A.; Barlow, P. N. ; Hulme, A. N.; Walkinshaw, M. D. ; Baldwin, A. J. ; Michel, J.
Chem. Sci. , 11, 2670-2680, 2020 doi:10.10139/c9sc04696h [bioRxiv] PDB 6GS6 BMRB 12023
BioSimSpace: An interoperable Python framework for biomolecular simulation
Hedges, L. O. ; Mey, A. S. J. S. ; Laughton, C. A. ; Gervasio, F. L. ; Mulholland, A. J. ; Woods, C. J. ; Michel, j. Journal of Open Source Software, 4(43), 1831, 2019 doi:10.21105/joss.01831

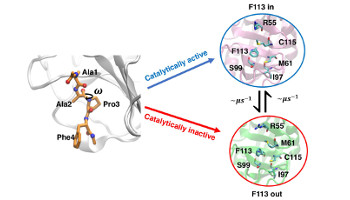
Allosteric effects in a catalytically impaired variant of the enzyme Cyclophilin A are unrelated to millisecond time scale motions
Wapeesittipan, P. ; Mey, A. S. J. S. ; Walkinshaw, M. D. ; Michel, J. Commun. Chem. 2: 41, 2019 doi:10.1038/s42004-019-0136-1 [bioRxiv]
Effect of set up protocols on the accuracy of alchemical free energy calculation over a set of ACK1 inhibitors
Granadino-Roldan, J. M. ; Mey, A. S.J.S.;Perez, J. J. ; Bosisio, S. ; Rubio-Martinez, J. ; Michel, J.
PLOS ONE 14(3): e0213217,2019 doi:10.1371/journal.pone.0213217 [bioRxiv]
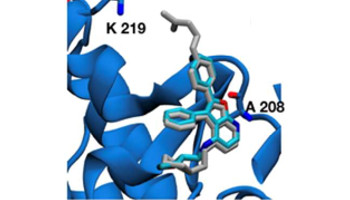
A computationally designed binding mode flip leads to a novel class of potent tri-vector cyclophilin inhibitors
De Simone, A. ; Georgiou, C. ; Ioannidis, H. ; Gupta, A. G. ; Juarez-Jimenez, J. ; Doughty-Shenton, D. ; Blackburn, E. A. ; Wear, M . A. ; Richards, J. P. ; Barlow, P. N. ; Carragher, N. ; Walkinshaw, M. D. ; Hulme, A. N. ; Michel, J.
Chem. Sci. , 10, 542- 547, 2019, doi:10.1039/C8SC03831GPDB: 6GJJ, 6GJM, 6GJY, 6GJP, 6GJI, 6GJR, 6GJL, 6GJN

Reproducibility of Free Energy Calculations Across Different Molecular Simulation Software
H. H. Loeffler, H. H. ; Bosisio, S. ; Ramos Matos G. D. ; Suh D. ; Roux, B. ; Mobley D. L. ; Michel, J.
J. Chem. Theory Comput. , 14, 5567- 5582, 2018, doi:10.1021/acs.jctc.8b00544 [chemRxiv]

Biomolecular simulations: from dynamics and mechanisms to computational assays of biological activity
Huggins, D. J. ; Biggin, P. C. ; Dämgen, M. A. ; Essex, J. W. ; Harris, S. A. ; Henchman, R. H. ; Khalid, S. ; Kuzmanic, A. ; Laughton, C. A. ; Michel, J; Mulholland, A. J. ; Rosta, E. ; Sansom, M. S. P. ; van der Kamp, M. W.
Wiley Interdisciplinary Reviews: Computational Molecular Science, 2018 , doi.org/10.1002/wcms.1393

Blinded Predictions of Standard Binding Free Energies: Lessons Learned from the SAMPL6 Challenge
Papadourakis, M. ; Bosisio, S. ; Michel, J.
J Comput. Aided Mol. Design, 32, 1047-1058, 2018, doi.org/10.1007/s10822-018-0154-6 [chemRxiv]

Metabolism and hydrophilicity of the polarised ‘Janus face’ all-cis tetrafluorocyclohexyl ring, a candidate motif for drug discovery
Rodil, A. ; Bosisio, S. ; Ayoup, M. S. ; Quinn, L. ; Cordes, D. B. ; Slawin, A. M. Z. ; Murphy, C. ; Michel, J. ; O'Hagan, D.
Chem. Sci. , 9, 3023-3028, 2018, doi:10.1039/C8SC00299A

Impact of domain knowledge on blinded predictions of binding energies by alchemical free energy calculations
Mey, A. S. J. ; Juárez-Jiménez, J. ; Michel, J.
J Comput. Aided Mol. Design, 32:199–210, 2018, doi:10.1007/s10822-017-0083-9 [bioRxiv]
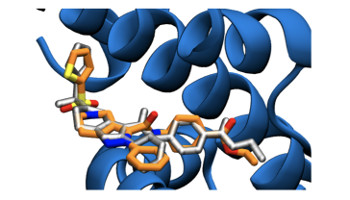
Pushing The Limits Of Detection Of Weak Binding Using Fragment Based Drug Discovery: Identification Of New Cyclophilin Binders

Assessment of Hydration Thermodynamics at Protein Interfaces with Grid Cell Theory
Gerogiokas, G. ; Southey, M. W. Y. ; Mazanetz, M. P. ; Heifetz, A. ; Bodkin, M. ; Law, R. J. ; Henchman, R. H. H. ; Michel, J.
J. Phys. Chem. B, 120(40), 10442-10452, 2016 doi:10.1021/acs.jpcb.6b07993

Blinded predictions of distribution coefficients in the SAMPL5 challenge
Bosiso, S ; Mey, A. S. J. S. ; Michel, J.
J. Comput. Aided Mol. Des., 30:1101-1114, 2016 doi:10.1007/s10822-016-9969-1

Blinded predictions of host-guest standard free energies of binding in the SAMPL5 challenge
Bosiso, S ; Mey, A. S. J. S. ; Michel, J.
J. Comput. Aided Mol. Des., 31:61-70, 2017 doi:10.1007/s10822-016-9933-0 [ PDF ]

Blinded predictions of binding modes and energies of HSP90-alpha ligands for the 2015 D3R Grand Challenge
Mey, A. S. J. S. ; Juárez-Jiménez J. ; Hennessy, A. ; Michel, J.
Bioorg. Med. Chem., 24(20), 4890-4899, 2016 doi:10.1016/j.bmc.2016.07.044 [ PDF ]

Elucidation of Non-Additive Effects in Protein-Ligand Binding Energies: Thrombin as a Case Study
Calabro, G. ; Woods, C. J. ; Powlesland, F. ; Mey, A. S. J. S.; Mulholland, A. J. ; Michel, J.
J. Phys. Chem. B, 120 (24), 5340-5350, 2016 doi:10.1021/acs.jpcb.6b03296 [ PDF ]

Impact of Ser17 phosphorylation on the conformational dynamics of the oncoprotein MDM2
Bueren-Calabuig, J. A. ; Michel, J.
Biochemistry, 55 (17), 2500-2509, 2016 doi:10.1021/acs.biochem.6b00127 [ PDF ]

FESetup: Automating Setup for Alchemical Free Energy Simulations
Loeffler, H. H. ; Michel, J. ; Woods, C. J.
J. Chem. Inf Model., 55 (12), 2485-2490, 2015 doi:10.1021/acs.jcim.5b00368
Evaluation of Selected Classical Force Fields for Alchemical Binding Free Energy Calculations of Protein-Carbohydrate complexes
Sushil K. Mishra, S. K. ; Calabró, G. ; Loeffler, H. H. ; Michel, J.. ; Koca J.
J. Chem. Theory Comput., 11 (7), 3333-3345, 2015 doi:10.1021/acs.jctc.5b00159 [ PDF ]

Elucidation of Ligand-Dependent Modulation of Disorder-Order Transitions in the Oncoprotein MDM2
Bueren-Calabuig, J. A. ; Michel, J.
PLoS Comput. Biol. , 11(6): e1004282, 2015 doi:10.1371/journal.pcbi.1004282
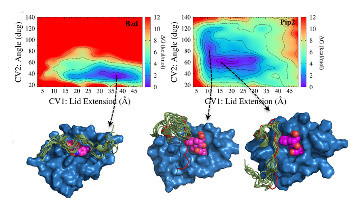
A Collective Variable for the Rapid Exploration of Protein Druggability
Cuchillo, R. ; Pinto-Gil, K. ; Michel, J.
J. Chem. Theory Comput., 11 (3), 1292-1307, 2015 doi:10.1021/ct501072t [ PDF ]
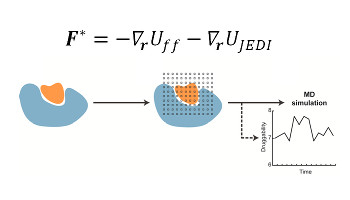
Evaluation of Water Displacement Energetics in Protein Binding Sites with Grid Cell Theory
Gerogiokas, G. ; Southey, M. W. Y. ; Mazanetz, M. P. ; Heifetz, A. ; Bodkin, M. ; Law, R. J. ; Michel, J.
Phys. Chem. Chem. Phys., 17, 8416-8426, 2015 doi:10.1039/C4CP05572A [ PDF
[ PDF
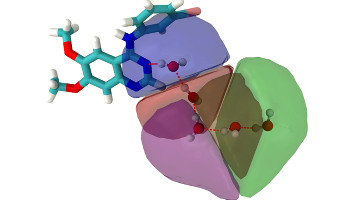
Evaluation of Host/Guest Binding Thermodynamics of Model Cavities with Grid Cell Theory
Michel, J. ; Henchman, R.H. ; Gerogiokas, G. ; Southey, M. W. Y. ; Mazanetz, M. P. ; Law, R. J. ;
J. Chem. Theory Comput., 10 (9), 4055-4068, 2014 doi:10.1021/ct500368p [ PDF ]
[ PDF ]

Strategies to calculate water binding free energies in protein-ligand complexes
Bodnarchuk, M. S. ; Viner, R. ; Michel, J. ; Essex, J. W.
J. Chem. Inf. Model., 54 (6), 1623-1633, 2014 doi:10.1021/ci400674k
Rapid Decomposition and Visualisation of Protein-Ligand Binding Free Energies by Residue and by Water
Woods, C. J. ; Malaisree, M. ; Michel, J. ; Long, B. ; McIntosh-Smith, S. ; Mulholland, A. J.
Faraday Discuss., 169, 477-499 2014 doi:10.1039/C3FD00125C
Current and Emerging Opportunities for Molecular Simulations in Structure-Based Drug Design
Michel, J.
Phys. Chem. Chem. Phys., 16, 4465-4477, 2014 doi:10.1039/C3CP54164A

Prediction of Small Molecule Hydration Thermodynamics with Grid Cell Theory
Gerogiokas, G. ; Calabro, G. ; Henchman, R.H. ; Southey, M. W. Y. ; Law, R. J. ; Michel, J.
J. Chem. Theory Comput., 10 (1), 35 - 48, 2014 doi:10.1021/ct400783h
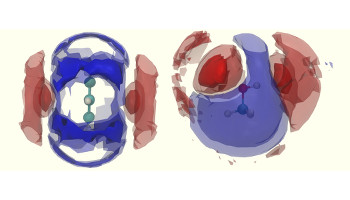
Luminescent, Enantiopure, Phenylatopyridine Iridium-Based Coordination Capsules
Chepelin, O.; Ujma, J.; Wu, X.; Slawin, A.; Pitak, M. B.; Coles, S., J.; Michel, J.; Jones, A. C.; Barran, P. E.; Lusby, P. J.
J. Am. Chem. Soc., 134 (47), 19334-19337, 2012 doi:10.1021/ja309031h
Mechanisms of small-molecule binding to intrinsically disordered proteins
Cuchillo, R.; Michel, J.
Biochem. Soc. Trans., 40 (5), 1004 -1008, 2012 doi:10.1042/BST20120086
The Impact of Small Molecule Binding on the Energy Landscape of the Intrinsically Disordered Protein c-Myc
Michel, J. ; Cuchillo, R.
PLoS ONE, 7 (7): e41070, 2012 doi:10.1371/journal.pone.0041070
Targeting the von Hippel–Lindau E3 Ubiquitin Ligase Using Small Molecules To Disrupt the VHL/HIF-1a Interaction
Buckley, D. L. ; Van Molle, I. ; Gareiss P. C. ; Seop Tae H. ; Michel, J. ; Noblin, D. J. ; Jorgensen, W. L. ; Ciulli, A. ; Crews, C. M.
J. Am. Chem. Soc., 134 (10), 4465 -4468, 2012 doi:10.1021/ja209924v
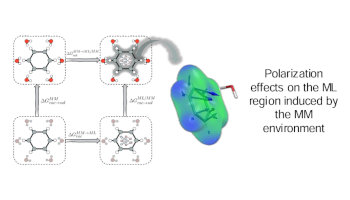
A Simple QM/MM Approach for Capturing Polarization Effects in Protein-Ligand Binding Free Energy Calculations
Beierlein, F. B. ; Michel, J. ; Essex, J. W.
J. Phys. Chem. B, 115 (17), 4911 -4926, 2011 doi:10.1021/jp109054j
Effects of Water Placement on Predictions of Binding Affinities for p38 MAPalpha Kinase Inhibitors
Lucarelli, J. ; Michel, J. ; Tirado-Rives, J. ; Jorgensen W. L.
J. Chem. Theory Comput., 6 (12), 3850 -3856, 2010 doi:10.1021/ct100504h
A Remote Arene Binding Site on Prostate Specific Membrane Antigen Revealed by Antibody-Recruiting Small Molecules
Zhang, A. X. ; Murelli, R. P. ; Barinka, C. ; Michel, J. ; Cocleaza, A. ; Jorgensen, W. L. ; Lubkowski, J. ; Spiegel, D. A.
J. Am. Chem. Soc., 132 (36), 12711 -12716, 2010 doi:10.1021/ja104591m
Rigorous free energy calculations in structure-based drug design
Michel, J. ; Foloppe, N. ; Essex, J. W.
Mol. Inf., 29, 570 - 578, 2010 doi:10.1002/minf.201000051
Prediction of protein-ligand binding affinity by free energy simulations: assumptions, pitfalls and expectations
Michel, J. ; Essex, J. W.
J. Comput. Aided Mol. Des., 24 (8), 639 -658 2010 doi:10.1007/s10822-010-9363-3
Bridged beta3-Peptide Inhibitors of p53-hDM2 Complexation - Correlation Between Affinity and Cell Permeability
Bautista, A. D. ; Appelbaum, J. S. ; Craig, C. J. ; Michel, J. ; Schepartz, A.
J. Am. Chem. Soc., 132 (9), 2904 -2906, 2010 doi:10.1021/ja910715u
Coarse-grain modelling of DMPC and DOPC lipid bilayers
Orsi, M. ; Michel, J. ; Essex, J. W.
J. Phys.: Condens. Matter, 22 (15), 155106, 2010 doi:10.1088/0953-8984/22/15/155106
Chemical Control Over Immune Recognition: A Class of Antibody-Recruiting Small Molecules (ARMs) that Target Prostate Cancer
Murelli, R. P. ; Zhang, A. X. ; Michel, J. ; Jorgensen W. L. ; Spiegel, D. A.
J. Am. Chem. Soc., 131 (47), 17090 -17092, 2009 doi:10.1021/ja906844e
Energetics of displacing water molecules from protein binding sites: consequences for ligand optimization
Michel, J. ; Tirado-Rives, J. ; Jorgensen W. L.
J. Am. Chem. Soc., 131 (42), 15403 -15411, 2009 doi:10.1021/ja906058w
Prediction of the water content in protein binding sites
Michel, J. ; Tirado-Rives, J. ; Jorgensen W. L.
J. Phys. Chem. B,113 (40), 13337 -13346, 2009 doi:10.1021/jp9047456
In silico improvement of b3-peptide inhibitors of p53•hDM2 and p53•hDMX
Michel, J. ; Harker, E. A. ; Tirado-Rives, J. ; Jorgensen W. L. ; Schepartz, A.
J. Am. Chem. Soc., 131 (18), 6356 -6357, 2009 doi:10.1021/ja901478e
Hit identification and binding mode predictions by rigorous free energy simulations
Michel, J. ; Essex, J. W.
J. Med. Chem., 51 (21), 6654 -6664, 2008 doi:10.1021/jm800524s
Prediction of partition coefficients by multiscale hybrid atomic level/coarse-grain simulations
Michel, J. ; Orsi, M ; Essex, J. W.
J. Phys. Chem. B, 112 (3), 657-660, 2008 doi:10.1021/jp076142y
Protein-ligand complexes: computation of the relative free energy of different scaffolds and binding modes
Michel, J. ; Verdonk, M.L ; Essex, J. W.
J. Chem. Theory Comput., 3 (5), 1645 -1655, 2007 doi:10.1021/ct700081t
Protein-ligand binding affinity predictions by implicit solvent simulations: a tool for lead optimization?
Michel, J. ; Verdonk, M.L ; Essex, J. W.
J. Med. Chem., 49 (25), 7427 -7439, 2006 doi:10.1021/jm061021s
The use of free energy simulations as scoring functions
Michel, J.
PhD thesis, University of Southampton, 2006 pdf
Efficient generalized Born models for Monte Carlo simulations
Michel, J. ; Taylor, R. D. ; Essex, J. W.
J. Chem. Theory Comput., 2 (3), 732 -739, 2006 doi:10.1021/ct600069r
The parameterization and validation of generalized Born models using the pairwise descreening approximation
Michel, J. ; Taylor, R. D. ; Essex, J. W.
J. Comput. Chem., 25, 1760-1770, 2004 doi:10.1002/jcc.20105
Patents
Cyclophilin Modulators
Michel, Julien.; Hulme, Alison, N. ; Doughty-Shenton, D. ; Kouridaki, M.
PCT Int. Appl. (2026), WO 2026/027787
Compounds for the modulation of cyclophilins function
Michel, Julien.; De Simone, Alessio ; Ioannidis, Harris ; Juarez-Jimenez, Jordi ; Georgiou, Charis ; Gupta, Arun ; Hulme, Alison, N. ; Walkinshaw, Malcolm, D. ; Doughty-Shenton, D.
PCT Int. Appl. (2020), WO 2020/043831A1
Compounds & methods for the inhibition of VCB E3 ubiquitin ligase
Crews, Craig, M.; Buckley, Dennis; Ciulli, Alessio; Jorgensen, William; Gareiss, Peter, C.; Molle, Inge, Van; Gustafson, Jeffrey; Tae, Hyun-Seop ; Michel, Julien ; Hoyer, Dentin, Wade; Roth, Anke, G.; Harling, John, David; Smith, Ian Edward, David; Miah, Afjal, Hussain; Campos, Sebastien, Andre; Le, Joelle
PCT Int. Appl. (2013), WO 2013/106646
Compounds & methods for the enhanced degradation of targeted proteins & other polypeptides by an E3 ubiquitin ligase
Crews, Craig, M.; Buckley, Dennis; Ciulli, Alessio; Jorgensen, William; Gareiss, Peter, C.; Molle, Inge, Van; Gustafson, Jeffrey; Tae, Hyun-Seop ; Michel, Julien ; Hoyer, Dentin, Wade; Roth, Anke, G.; Harling, John, David; Smith, Ian Edward, David; Miah, Afjal, Hussain; Campos, Sebastien, Andre; Le, Joelle
PCT Int. Appl. (2013), WO 2013/106643