
Our Research
Protein dynamics
Internal motions are essential for most proteins to carry out their biological function. A range of molecular simulation methodologies are used to generate atomic level resolution models of protein dynamics from pico to millisecond timescales. Detailed analyses of the resulting models is pursued to understand how protein dynamics is influenced by environmental factors (ligands, mutations), and to elucidate protein allostery mechanisms.
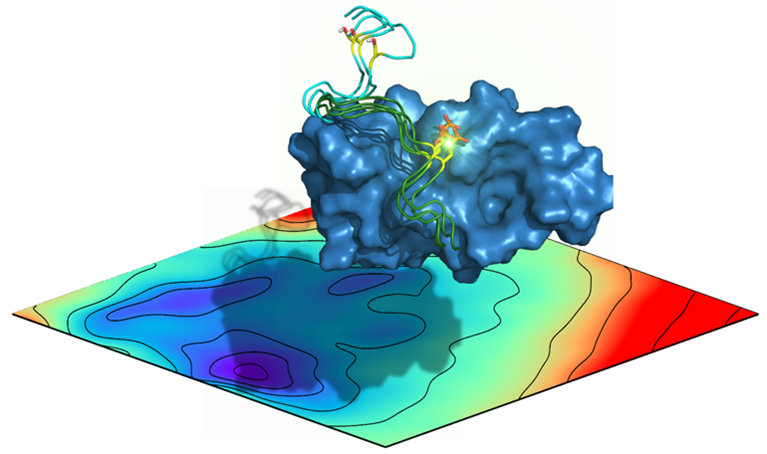
Impact of phosphorylation on the free energy landscape of the MDM2 disordered lid region.
Molecular simulations-driven ligand design
The group develops and applies molecular simulation methodologies grounded in statistical thermodynamics to estimate ligand properties that are relevant for early-stage drug discovery efforts (binding affinity, selectivity and thermodynamic signatures, partition coefficients). Robust implementation is sought by following modern software engineering practices. The overall objective is to enable routine and high-throughput use of biomolecular simulations for drug discovery efforts.

Binding energy estimates for thrombin ligands
Ensemble-based Drug Design
Molecular simulations are used to generate representations of protein conformational ensembles, and to guide the design of ligands that preferentially bind to selected protein conformations to achieve the desired functional effect. Hypotheses are tested via organic syntheses and biophysical measurements. Through the rational modulation of protein conformational ensembles we aim to open up new opportunities for the discovery of potent and selective ligands for proteins that challenge conventional drug discovery methods.
Simulated dynamics of a cyclophilin-fragment complex